предыдущая страница
следующая страница
5.3. Исследование комплексных соединений фуллеренов методом рентгеновской фотоэлектронной спектроскопии и спектроскопии потерь энергии электронов
Известно, что поверхность твердого тела по своему составу, как правило, отличается от объемного состава. По этой причине при использовании поверхностно-чувствительных методов для исследо-вания электронных свойств твердого тела всегда должны сопоставляться глубина зоны анализа и размер приповерхностной области, где отличия поверхности от объема существенны. Естественно ожидать, что для кристаллов фуллерена, где молекулярные взаимодействия являются слабыми, отличием поверхности от объема можно пренебречь. Это было продемонстрировано на примере угловой зависимости спектров потерь энергии электронов. Однако не для всех соединений на основе фуллеренов ситуация такая же. Далее будут приведены примеры, где поверхность молекулярных кристаллов отличается от объема. Будут обсуждены причины этих различий. Для случая, когда различиями объема и поверхности можно пренебречь, описывается пример использования рентгеновской фотоэлектронной спектроскопии для исследования электронных взаимодействий в фуллеридах.
5.3.1. Об отличии поверхности и объема для кристаллов молекулярных комплексов состава (D)x•(Ф)•(R)y
В настоящее время получено значительное количество соединений фуллеренов, заметную долю среди которых занимают молекулярные комплексы (D)x•(Ф)•(R)y, где Ф - фуллерен С60 или С70;
D - донор; R - растворитель. Доноры могут быть как неорганическими (S8 [1], S4N4 [2]), так и органическими (BEDT-TTF [3, 4], OMTTF [3], BEDO-TTF [5], BNDY [6], BTX [7, 8]). Эти молекулярные комплексы характеризуются малой степенью ионности, что выражается в небольшом (в пределах 2 см-1) смещении полос поглощения в их
ИК-спектрах, обусловленных колебаниями атомов молекулы фуллерена [1, 2, 5, 9]. Результаты исследований таких соединений методом рентгеновской фотоэлектронной спектроскопии также свидетельствуют о небольшом перераспределении зарядов при комплексообразовании [10, 11]. Для некоторых комплексов была установлена кристаллическая структура [1, 8] и отмечено незначительное изменение геометрии
фуллереновой компоненты, что подтверждает представление об отсутствии заметного переноса заряда в соединениях такого рода.
Анализ отношения интегральных интенсивностей пиков S2p и C1s в рентгеновских фотоэлектронных спектрах поликристаллов С60•2S8 показал [12], что поверхность образца в условиях высокого вакуума и облучения рентгеновскими квантами обедняется серой. Так, отношение [S/C60], определенное из РФ-спектров, оказалось равным 11 и 5 для свежеприготовленного образца и образца после длительного пребывания в камере спектрометра. В кристаллах, образующихся в системе фуллерен-тетраселенотетрацен-сероуглерод, также обнаружено обеднение приповерхностных слоев серой вследствие десорбции сероуглерода в высоком вакууме [13]. Эти случаи разного состава приповерхностного слоя и объема, в принципе, можно объяснить экспериментальными условиями.
На рис. 5.8 представлены спектры Te3d для кристаллов состава ВТХ•С70•CS2 [14]. Там же для сравнения приведены спектры чистого донора и кристаллов ВТХ•С60•CS2. Видно, что спектр ВТХ•С60•CS2 близок к таковому для чистого донора (сдвиг пика Te3d5/2 не превышает 0.4 эВ). По положению этого пика в доноре (583.8 эВ) и в кристаллах (584.2 эВ) состояние атома теллура следует описать как Te2+. В кристаллах ВТХ•С70•CS2 помимо пиков Te2+ отчетливо появляются пики Te4+ (сдвиг в сторону более высоких энергий связи на 2.2 эВ). В то же время для комплекса ВТХ•С70•CS2 был проведен рентгено¬структурный анализ, в результате которого установлено, что конформация донора ВТХ в нем не отличается от таковой в кристаллах ВТХ•С60•CS2 [7]. В ИК-спектрах кристаллов ВТХ•С60•CS2 и ВТХ•С70•CS2 также не было отмечено существенной разницы в положении полос поглощения, относящихся к колебаниям атомов донора [2]. Следовательно, зафиксированное состояние Te4+ относится к тонкому приповерхностному слою и его суммарная концентрация достаточно мала, чтобы быть обнаруженной в ИК-спектрах.
На рис. 5.9 представлен РФ-спектр S2p-кристаллов (BNDY)•(C60) [14]. Спектр состоит из двух пиков - основного с энергией связи Есв, равной 164.1 эВ, и дополнительного с Есв = 169,8 эВ. Положение дополнительного пика соответствует состоянию S6+ [11]. Отметим, что дополнительный пик отсутствует в спектре исходного BNDY и BNDY из контрольного опыта (кристаллизация из бензола в условиях роста кристаллов (BNDY)•(C60)).
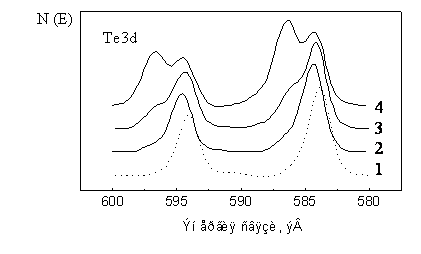
Рис. 5.8. РФ-спектры Te3d кристаллов БТХ (1), BTX•C60•CS2 (2) и BTX•C70•CS2 (3, 4) после вычитания линейного фона и нормировки пиковой интенсивности. Спектры 3 и 4 характеризуют поверхность кристаллов BTX•C70•CS2 из разных синтезов.
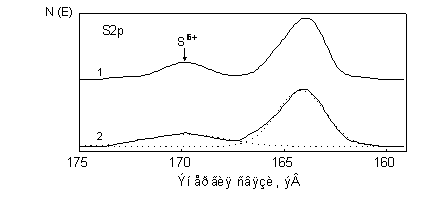
Рис. 5.9. РФ-спектры S2p поверхности кристаллов (BNDY)•(C60) (1) и (BEDO-TTF)•C60•C6H6 (2). Точками показана разложение спектра на гауссовы компоненты.
Дополнительный пик с Есв (S2p) = 169.8 эВ был также обнаружен на РФ-спектре кристаллов (BEDO-TTF)•C60•C6H6 [14]. Основной пик в этом случае находился при Есв = 164.2 эВ. В то же время на спектрах чистого донора и комплекса (BEDO-TTF)2•C60, который был получен при использовании в качестве растворителя CS2, пика, который можно было бы приписать S6+, обнаружено не было.
Отметим, что эффект окисления наблюдается только для незначительного числа доноров. Так, например, не было обнаружено присутствия серы в высоких степенях окисления на поверхности кристаллов (DB-TTF)•C60•C6H6, (DB-TTF)•C70•C6H6, (EDY-BEDT-DT)•C60•C6H6, (TMDTDM-TTF)2•C60•(CS2)3 и др., где (DB-TTF) - дибензотетратиафульвален; (EDY-BEDT-DT) - 2,2’-этандиилиден-бис(4,5-этилендитио-1,3-дитиол), (TMDTDM-TTF) – тетраметиленди-тиодиметилтетратиафульвален [11].
Таким образом, для соединений фуллеренов и таких доноров, как BTX, BNDY, BEDO-TTF и S4N4, наблюдается одинаковый эффект - на поверхности кристаллов (D)x•(Ф)•(R)y часть гетероатомов находится в высокоокисленном состоянии. Как правило, индивидуальный донор (BTX, BNDY, BEDO-TTF) достаточно устойчив к действию кислорода при длительном хранении на воздухе в нормальных условиях или при медленной кристаллизации в условиях получения комплекса. Более того, для некоторых доноров (S4N4) кристаллизация в условиях получения соответствующего комплекса с С60 может приводить к его восстановлению. Появление высокоокисленных состояний донора на поверхности кристаллов (D)x•(Ф)•(R)y обусловлено присутствием фуллерена, который играет роль катализатора и/или переносчика активного кислорода. Координация донора с молекулой фуллерена, на которой адсорбирован кислород, может способствовать окислению донора уже при комнатной температуре. Другой причиной окисления донора может быть его активация за счет донорно-акцепторного взаимодействия в комплексе. В этом случае большое влияние на процесс окисления будет иметь структура комплекса.
5.3.2. Сильное взаимодействие пи-электронов C60- и фенильных групп в фуллеридах [PPh4]2[C60][X], где Ph = C6H5, X = Cl, Br или I [15]
Фуллериды тетрафенилфосфония являются одним из немногочисленных примеров достаточно устойчивых на воздухе твердых тел, где молекула C60 находится в восстановленной форме
[16-18]. Для кристаллической структуры этих соединений характерны полная изоляция ионов C60 в очень симметричном окружении с укороченными С---С-контактами между С60 и фенильными группами.
Известно, что для твердого фуллерена С60 энергия связи электронов С1s равна 285.0 эВ [8]. Очевидно, что переход С60 - С60- не может существенно изменить значение Есв(C1s), поскольку избыточный электрон распределяется между 60-ю атомами углерода. Следовательно, в РФ-спектрах исследуемых солей пик C1s от атомов углерода фуллерена должен также находиться при Есв = 285.0 эВ.
Пик С1s от трифенилфосфинового лиганда также наблюдается при 285.0 эВ и его положение практически не зависит от заряда переходного металла в комплексе с этим лигандом (см., например, [20]). Можно полагать, что и для иона [PPh4]+ величина Есв(C1s) также не будет отличаться от 285.0 эВ. Подтверждение этого предположения можно найти в табл. 5.3, где приведены значения полуширины фотоэлектронного пика С1s (дельтаC1s) для исследуемых соединений и соединений сравнения. Видно, что значение дельта C1s для солей [PPh4]2[C60][X] только на 0.3 эВ превышает таковое для твердого С60. Таким образом, для изучаемых соединений калибровку спектров с высокой степенью точности можно проводить по внутреннему стандарту (Есв(C1s) = 285.0 эВ).
Таблица 5.3. Параметры РФ-спектров (эВ). Обозначения см. в тексте.
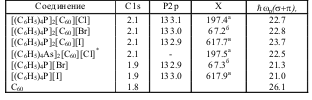
Как следует из табл. 5.3, при переходе от [PPh4][X] к [PPh4]2[C60][X] значение Есв(пи2p) остается постоянным в пределах экспериментальной точности (плюс-минус 0.1 эВ). Следовательно, зарядовое состояние катиона в сравниваемых соединениях одинаково, а на молекуле фуллерена находится заряд -1, что соответствует данным, полученным ранее [16-18]. Положение РФ-линий анионов Х- также не
меняется при переходе от солей, не содержащих фуллерена, к фуллеренсодержащим, что также соответствует ионному строению соединений [PPh4]2[C60][X].
Для соединений с PPh3-лигандами вблизи основного пика C1s со стороны более высоких энергий связи наблюдается характерный пик, появление которого обычно связывают с возбуждением перехода в фенильном кольце. Вероятность этого перехода, измеренная как отношение интенсивности сателлитного пика к интенсивности основного пика, равна 0.06, а энергия (т. е. расстояние между максиму-мами основного и сателлитного пиков) равна 6.7 эВ [21]. На
РФ-спектрах солей типа [PPh4][X], где X = Cl, Br или I, наблюдается такой же сателлит, причем его энергия и интенсивность практически совпадают с таковыми для PPh3.
Для солей [PPh4]2[C60][X] интенсивность сателлита, сопровождающего пик С1s, существенно уменьшается, а максимум сдвигается в сторону основного пика (рис. 5.10). Можно полагать, что причина этого явления заключается в наложении спектров С1s от катиона [PPh4]+ и аниона [C60]-. Однако синтез спектра С1s путем сложения спектров от [PPh4][X] и C60 в пропорции, равной отношению атомов углерода катиона и аниона в [PPh4]2[C60][X] (48 : 60), дает сателлитный пик с интенсивностью, в 2 раза превышающей таковую на экспериментальном спектре [PPh4]2[C60][X], и с максимумом, отстоящим от основного пика на 6.0 эВ. Таким образом, наблюдаемый эффект вызван либо тем, что пи-электроны фенильных колец и фуллерена выступают в этом процессе как одна единая система, либо тем, что при образовании С1s-дырки на атоме углерода фенильной группы возбуждение перехода оказывается менее выгодным по сравнению с возбуждением плазменных осцилляций пи-электронов C60-, энергия которых отличается от этой величины в случае твердого C60. Выбор между этими двумя механизмами пока не представляется возможным. Тем не менее, в любом случае упомянутый эффект однозначно указывает на то, что пи-электроны фенильных групп существенным образом скоррелированы с пи-электронами фуллерена. Следствием этого взаимодействия может быть высокая устойчивость исследуемых солей на воздухе. Согласно рентгеноструктурным данным [18], плоскость фенильного кольца, наиболее близко расположенного к ион-радикалу C60-, стерически блокирует наиболее активную двойную интерпентогональную связь аниона. Можно также предположить, что обнаруженное в настоящей работе взаимодействие пи-электронов фенильного кольца и приводит к уменьшению реакционной способности двойной связи С=С-аниона.
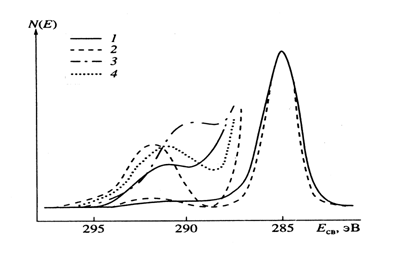
Рис. 5.10. РФ-спектры C1s от [PPh4]2[C60][Br] (сплошная линия) и сател-литная структура, сопровождающая пик С1s, для [PPh4]2[C60][Br] (1), [PPh4][Br] (3), C60 (2) и для смеси
С60 + [PPh4][Br] (4). О построении “смесевого” спектра см. в тексте. Все спектры нормированы на интенсивность пика C1s; cателлитная структура изображена в увеличенном по оси Y масштабе (х8).
Описанное отклонение измеренной величины от рассчитанной в случае С60 было объяснено локализацией плазменных колебаний валентных электронов. Уменьшение тетта в случае изучаемых соединений по сравнению с фуллереном обусловлено в первую очередь делокализацией плазменных осцилляций вследствие укороченных контактов между шарами C60 и фенильными группами катиона.
Литература
-
Roth G., Adelmann P. // Appl. Phys. A – 1993. – Vol. 56. – P. 169.
-
Конарев Д.В., Любовская Р.Н., Рощупкина О.С., Тарасов Б.П., Шульга Ю.М. // Изв. PАН. Cер. хим. – 1997. – P. 37.
-
Izuoka A., Tachikawa T., Sugawara T., Suzuki Y., Konno M., Saito Y., Shinohara H. // Chem. Commun. – 1992. – N 19. – P. 1472.
-
Saito G., Teramoto T., Otsuka A., Sugita Y., Ban T., Kusunoki M., and Sakagikhi K. // Synth. Metals. – 1994. – Vol. 64. – P. 350.
-
Konarev D.V., Lyubovskaya R.N., Roschupkina O.S., Shulga Yu.M., Valeev E.F., and Slovokhotov Yu.L. // Abstracts of ICSM’96. – P. 287.
-
Konarev D.V., Lyubovskaya R.N., Roschupkina O.S., Shulga Yu.M., Kaplunov M.G., Kremenskaya I.N., Rosenberg L.P., Hasanov S.S., Shibaeva R.P. // Molecul. Mater. – 1996. – 8, N 1-2. – P. 79-82.
-
Нарымбетов Б.Ж., Хасанов С.С., Зорина А.В., Розенберг Л.П., Шибаева Р.П., Конарев Д.В., Любовская Р.Н. // Кристаллография. – 1997. – 42, № 5. – P. 851-857.
-
Kveder V.V., Steinman E.A., Narimbetov B.Z., Khasanov S.S., Rozenberg L.P., Shibaeva R.P., Bazhenov A.V., Gorbunov A.V., Maksimuk M.Yu., Konarev D.V., Lyubovskaya R.N., and
Osipyan Yu.A. // Chem.Phys. – 1997. – Vol. 216 – P. 407.
-
Crane J.D., Hitchcock P.B., Kroto H.W., Taylor R., and Walton R.M. Chem.Commun. – 1992. – P. 1765.
-
Shul'ga Yu.M., Rubtsov V.I., Vasilets V.N., Lobach A.S., Spitsyna N.G., and Yagubskii E.B. // Synth. Metals. – 1995. – Vol. 70. – P. 1381.
-
Shulga Yu.M., Lyubovskaya R.N., and Konarev D.V. // Phys. Low-Dim. Struct. – 1997. – Vol. 1/2. – P. 103.
-
Шульга Ю.М., Рубцов В.И., Василец В.Н. и др. // Изв. PАН. Сер. хим. – 1994. – N 12. – C. 2149-2152
-
Лаухина Е.Э., Шульга Ю.М., Рубцов В.И. и др. // Изв. PАН. Cер. хим. – 1995. – N 5. – C. 845-848.
-
Шульга Ю.М., Любовская Р.Н., Конарев Д.В. // Журнал физической химии. – 1997. – 71, N 12. – C. 2179-2182.
-
Шульга Ю.М., Моравский А.П., Лобач А.С., Рубцов В.И. // Письма в ЖЭТФ. – 1992. – 55, N 2. – C. 137-140.
-
Bilow U., Jansen M. // Chem. Commun. – 1994. – P. 403.
-
Penicaud A., Perez-Benitez A., Gleason V.R. et. al. // J. Amer. Chem. Soc. – 1993. – Vol. 115. – P. 10392-10395.
-
Spitsyna N.G., Cristenko V.V., Shilov G.V. et. al. // Mol. Struct. – 1996 (in press).
-
Jost M.B., Troullier N., Poirier D.M. at. al. // Phys. Rev. – 1991. – B44, N 4. – P. 1966-1071.
-
Cook C.D., Wan K.Y., Gelius U. et. al. // J. Amer. Chem. Soc. – 1971. – Vol. 93. – P. 1904-1910.
-
Shul’ga Yu.M., Bulatov A.V., Gould R.A.T. et. al. // Inorg. Chem. – 1992. – Vol. 31. – P. 4704-4707.
предыдущая страница
следующая страница
|
